Telomeres shorten in heart muscle cells from people with Duchenne muscular dystrophy. A Stanford Medicine study finds blocking this process improves the health of these cells grown in a dish.
January 31, 2023 - By Krista Conger
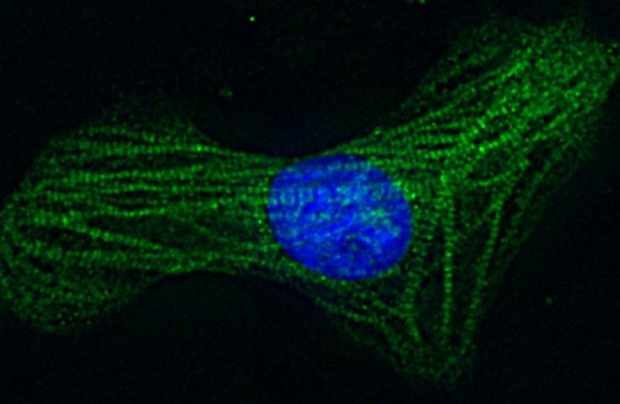
A healthy cardiomyocyte, or heart muscle cell. Stanford researchers have found that a protein that prevents telomeres from shortening helps cardiomyocytes from patients with Duchenne muscular dystrophy live longer.
Asuka Eguchi
A protein that prevents the shortening of telomeres — the protective caps on the ends of chromosomes — helps heart muscle cells derived from patients with Duchenne muscular dystrophy live longer and appear healthier in a laboratory dish, according to a study by researchers at Stanford Medicine.
The finding is notable as heart failure caused by loss of these cells, called cardiomyocytes, is the leading cause of death for people with the disease.
“One of the most unexpected findings in our previous research is that telomeres shorten over time in cardiomyocytes in the hearts of people with Duchenne muscular dystrophy,” said Helen Blau, PhD, professor of microbiology and immunology. “Now we show that we can prevent that shortening in a laboratory dish, and when we do so, the diseased cardiomyocytes begin to resemble those from healthy people. They also live longer.”
The cells’ dramatic turnaround was not anticipated.
“We had hoped to see an increase in cardiomyocyte longevity, based on our previous research,” said Asuka Eguchi, PhD, an instructor in the Stanford Cardiovascular Institute and a member of Blau’s lab. “But we were quite surprised to also see such an improvement in the appearance of the cells, both in their size and in the prevalence of the proteins that allow muscle cells to contract. Premature loss of cardiomyocytes leads to scarring and stiffening of the heart tissue and significantly impairs its function. Now we’re homing in on a strategy to combat this damage.”
Blau, who is also the Donald E. and Delia B. Baxter Foundation Professor and director of the Baxter Foundation Laboratory for Stem Cell Biology, is the senior author of the research, which was published online Jan. 31 in the Proceedings of the National Academy of Sciences. Eguchi is the lead author of the study.
A shortened life
About one in 5,000 boys are born with Duchenne muscular dystrophy, which is caused by a mutation in a gene for a protein called dystrophin. Dystrophin connects the inside of muscle cells to proteins and other molecules on structures outside the cell, enabling muscles to contract properly. Without dystrophin, the muscles progressively weaken, and patients usually die in young adulthood due to heart and breathing difficulties.
A cardiomyocyte from a person with Duchenne muscular dystrophy.
Asuka Eguchi
Telomeres are often thought of as the body’s molecular clock because they shorten slightly every time a cell divides. When they get too short, the cell stops dividing and dies. Until recently, however, they’ve been thought to be largely stable in cells like cardiomyocytes, which divide infrequently. But research from Blau’s lab in 2016 and 2018 found that people with Duchenne muscular dystrophy, as well as those with a type of heart failure called cardiomyopathy, have telomeres that are between 25% and 40% shorter than those in healthy people.
The abnormally shortened telomeres activate a damage response that interferes with the cells’ energy centers and hampers the heart’s ability to pump blood throughout the body, the researchers found. In this study, they sought ways to combat that.
“We wanted to see if we could re-create this telomere shortening in a laboratory dish and whether we might be able to prevent it,” Eguchi said.
The researchers used induced pluripotent stem cells generated from two people with Duchenne muscular dystrophy to make cardiomyocytes with mutations in the dystrophin gene. (Induced pluripotent stem cells can be made from skin or other cells collected painlessly from people and can be coaxed in a laboratory dish to become nearly any type of cell in the body.) They grew the cardiomyocytes in a laboratory dish and observed how they looked and functioned compared with healthy cells in which the dystrophin mutation had been genetically corrected in the laboratory. The cells were otherwise genetically identical.
They saw that the diseased cardiomyocytes were about 40% smaller than the cells in which the dystrophin mutation had been corrected. The patterns of troponin — a calcium-binding protein on the filaments that produce muscle contraction — were also less dense than those seen in healthy cells.
Chromosome guards
Telomeres are made up of the same building blocks, called nucleotides, of the genes they protect. But rather than coding for proteins, they contain a specific sequence of six nucleotides repeated hundreds or thousands of times on the ends of chromosomes. These repeats serve as a physical buffer, protecting the ends of the chromosomes from the shortening that naturally occurs during the DNA replication that precedes cell division. After a finite number of cell divisions, the shrinking telomeres trigger a state of cellular hibernation and eventual death — effectively limiting the lifespan of most human cells.
A muscular dystrophy cardiomyocyte exposed to the protein that protects the telomeres from shortening.
Asuka Eguchi
Telomeres, in turn, are protected by a group of several proteins that form a structure called shelterin, which guards against damage and regulates the activity of a telomere-lengthening protein, telomerase, that is active in stem cells and in some cancer cells.
The researchers found that exposing the diseased cardiomyocytes to TRF2, a shelterin protein, prevented the telomeres from shortening. It also increased the number of cells that survived more than 30 days.
“We saw these beautiful morphological changes,” Eguchi said, “as well as an increase in the density of troponin, a protein that is a key component of the filaments that make up the contractile apparatus. We are excited about the possibility of one day using a similar approach to protect telomeres in patients with Duchenne muscular dystrophy or perhaps even other types of cardiomyopathy.”
The researchers caution, however, that much more research needs to be conducted to learn whether overexpression of TRF2 would be safe and effective in people with these conditions. “This would have to be a carefully targeted gene therapy,” Eguchi said, “because there is evidence that exposing dividing cells to TRF2 can cause a lot of havoc in the body.”
A TRF2-based gene therapy also would not address the cause of Duchenne muscular dystrophy, which is the lack of the dystrophin protein throughout the body.
“This alone could not cure the disease,” Blau said. “But it could perhaps ameliorate the heart failure experienced by these patients. Ideally, we would one day be able to give a combination therapy that both restores the ability to produce dystrophin and helps maintain the viability of the cardiomyocytes.”
The study was funded by the National Institutes of Health (grants R01 HL126527, R01 HL130020, R01 HL123968, R01 HL146690 and R01 HL159340), the American Heart Organization, the Muscular Dystrophy Association, the California Institute for Regenerative Medicine, the Keck Foundation, the Baxter Foundation, the Li Ka Shing Foundation, the Tobacco-Related Disease Research Foundation, a Stanford School of Medicine dean’s postdoctoral fellowship, a Stanford Translational Research and Applied Medicine pilot grant, and the Stanford Bio-X Summer Undergraduate Research Program.
-
 Krista CongerKrista Conger is a senior science writer in the Office of Communications. Email her at kristac@stanford.edu.
Krista CongerKrista Conger is a senior science writer in the Office of Communications. Email her at kristac@stanford.edu.
About Stanford Medicine
Stanford Medicine is an integrated academic health system comprising the Stanford School of Medicine and adult and pediatric health care delivery systems. Together, they harness the full potential of biomedicine through collaborative research, education and clinical care for patients. For more information, please visit med.stanford.edu.

